Tutorial - Denoising, Membrane Segmentation and Annotation and Directional Picking¶
This tutorial covers a part of the full data processing pipeline in cryo electron tomography, concretely from the tomogram to the initial model generation after having picked the particles. All the data processing has been carried out in Scipion, using the plugins listed below for each step:
- Import tomograms - scipion-em-tomo
- Tomogram normalization - scipion-em-imod
- Tomogram denoising - scipion-em-jjsoft_
- Tomogram segmentation, annotation and tomomask (segmemtation) resizing - scipion-em-tomosegmemtv
- Directional picking (preseg, graphs, filaments and picking) - scipion-em-pyseg
- Remove duplicates (filter picked particles by distance) and filter by normal - scipion-em-tomo3d
- Extract particles - scipion-em-emantomo
- 2D classification and rot angle randomization - scipion-em-pyseg
- Generate an initial model - scipion-em-reliontomo
Thus, 8 different plugins will be used in this tutorial, highlighting the power of Scipion in terms of interoperability. Figure below shows and scheme of the main workflow steps proposed for this tutorials and the plugins used to carry them out.
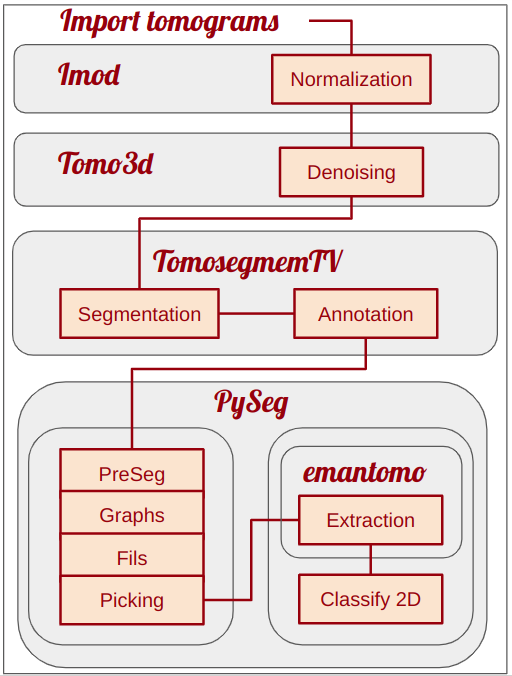
Table of Contents
- Tutorial - Denoising, Membrane Segmentation and Annotation and Directional Picking
- Objective
- Associated resources
- The dataset
- Preparing the project
- Importing the tomogram
- Tomogram normalization
- Tomogram denoising
- Membrane segmentation
- Membrane annotation
- Resize the tomomasks (segmentations)
- Directional picking with PySeg
- Picking post processing
- Subtomogram extraction
- 2D classification
- Extra material
Objective¶
The aim of this tutorial is to pick the membrane ribosomes of an in situ tomogram using segmentation, annotation and directional picking protocols inside Scipion framework.
Associated resources¶
Here you can find resources associated with this content, like videos or presentations used in courses and other documentation pages:
The dataset¶
PySeg presentation
The dataset reference used in this tutorial is EMD-10439, which consists of an in situ tomogram of intact P19 cells acquired with phase-plate, with a sampling rate of 13.68 Å/voxel and dimensions (X, Y, Z) = (928, 928, 500) pixels.
Preparing the project¶
First of all, open a terminal and execute the command scipion3 to run Scipion. After that:
- Click on button “Create Project”.
- Write a name for it. We’ll name it tomo_workshop_2021_tomosegmemTV_pyseg.
- Click on button “Create”.
Note: the protocols can be located on the left panel of the project interface or directly search via ctrl + f and typing the keywords that may represent what it is desired to be found, like a plugin name, a protocol name, an action, etc.
Importing the tomogram¶
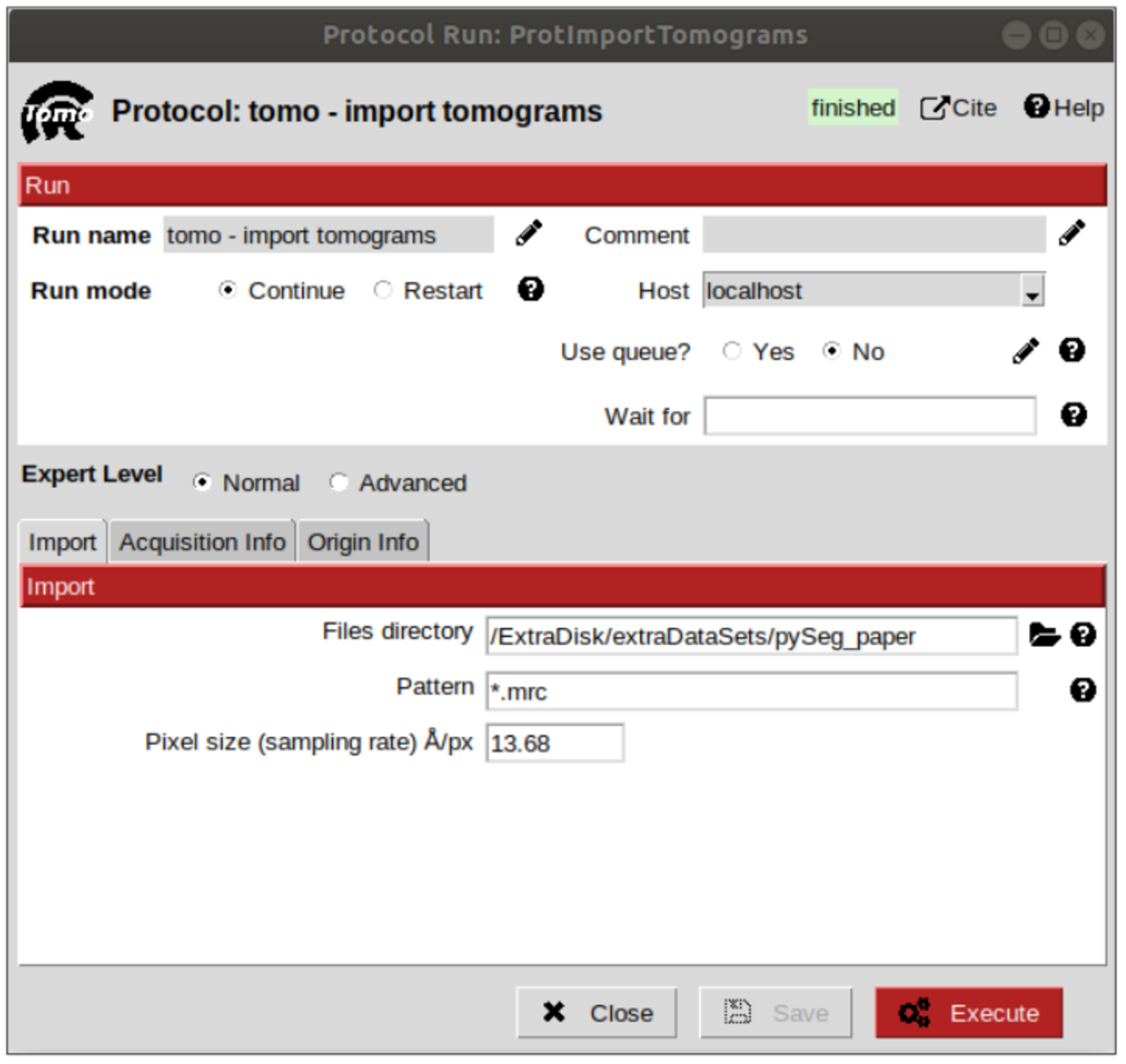
Let’s begin importing the tomogram. This is the entry point to Scipion, in which external data files are represented as Scipion objects, which is a common representation of the data used to make all the different packages speak to each other. To do that, simply look for a protocol named “import tomograms” and click on it. On tab “Import”, introduce the directory in which the tomogram file is located, then the full name or a pattern in the second field and finally the sampling rate, which is, as mentioned before, 13.68 Å/voxel. Leave the other two tabs with the default values and click on “Execute” button.
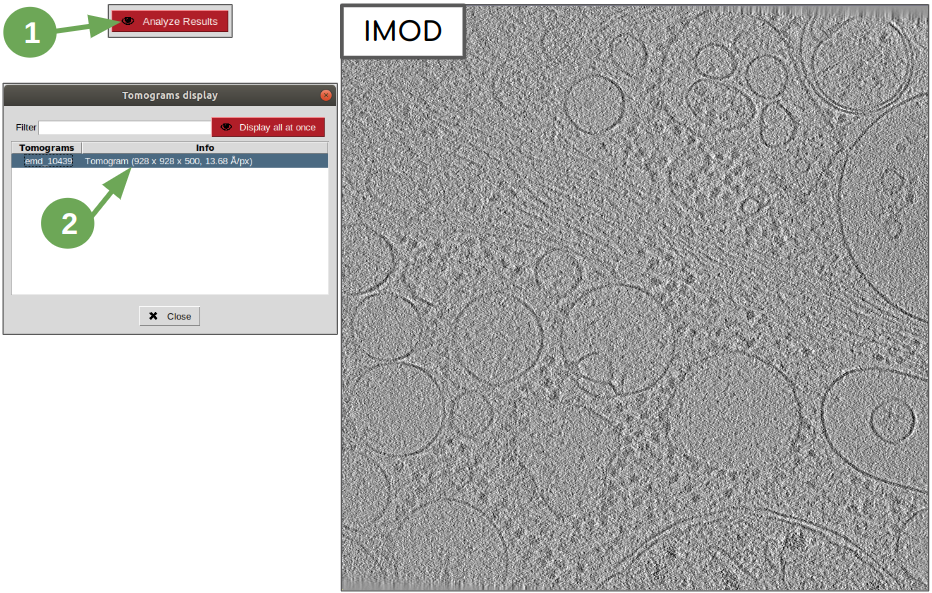
The imported data can be now visualized by clinking on button “Analyze”, located on the top right corner of the bottom panel. This will generate an auxiliary window which will lists the tomograms contained in the set imported. In our case, there is only one tomogram. To open it with IMOD’s viewer 3dmod (integrated as part of plugin scipion-em-imod), simply double click on it.
Tomogram normalization¶
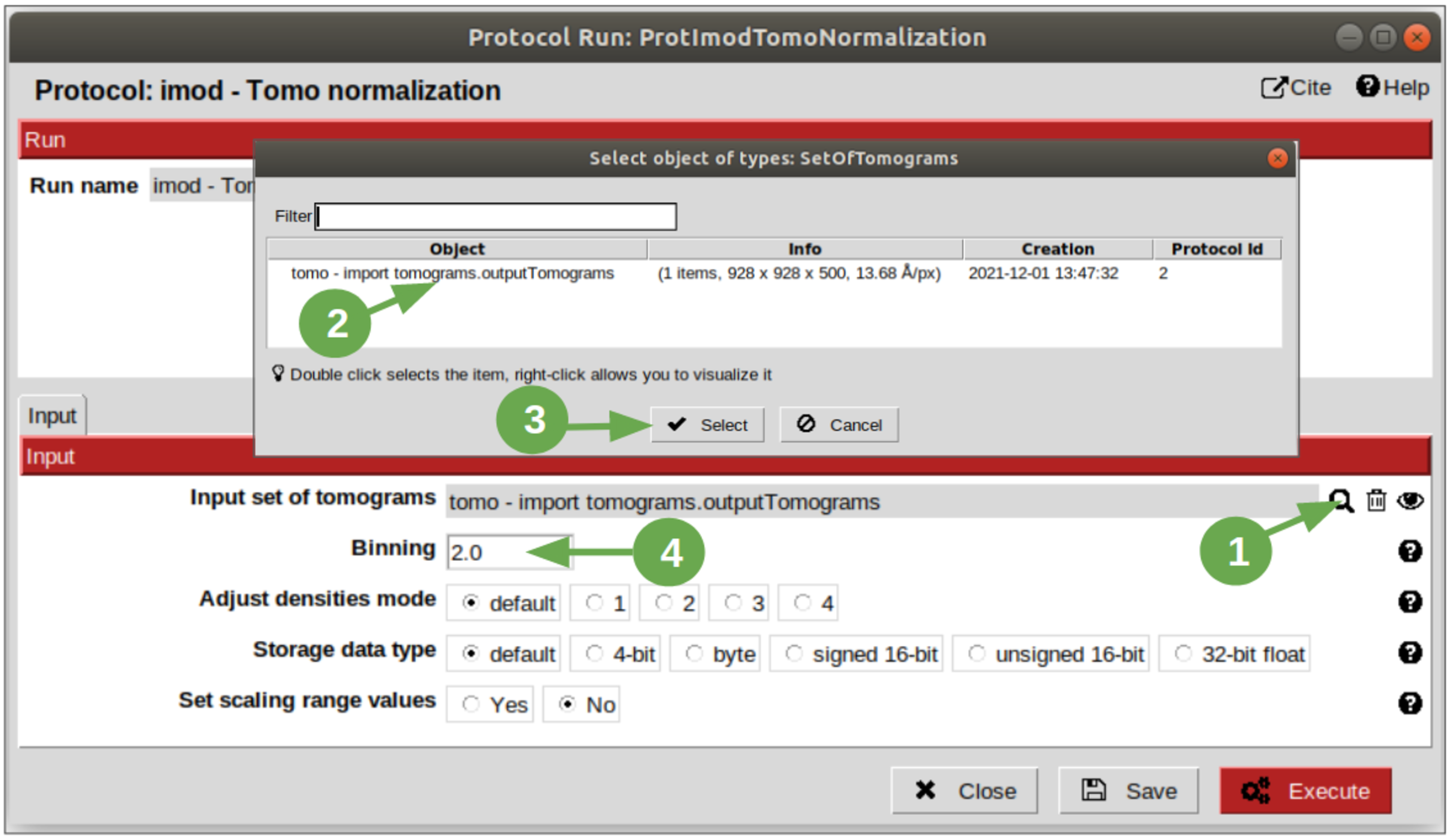
In this step, we are going to divide by two the size of the tomogram in order to make the denoising, segmentation and annotation steps faster and, in the case of the membranes segmentation and annotation, making it easier to the algorithm to detect them, because of the enhanced contrast as the binning gets higher. To do that, we are going to use the protocol called “tomo normalization” from plugin scipion-em-imod. Once the protocol form is on the screen, follow the steps listed below:
1. To get the pointer to the tomogram previously imported, click on the magnifier icon. This action will open an auxiliary window which will lists the existing objects of the same type as expected.
- At this point of the wokflow, we only have the tomogram imported before. Hence, select it.
- Click on “Select” button.
4. Introduce vale 2 in “Binning” field, to indicate that the resulting tomogram must be half of the size of the input tomogram. Consequently, the sampling rate of the output tomogram will be the double, as can be observed in the summary panel at the bottom of the project interface.
Tomogram denoising¶
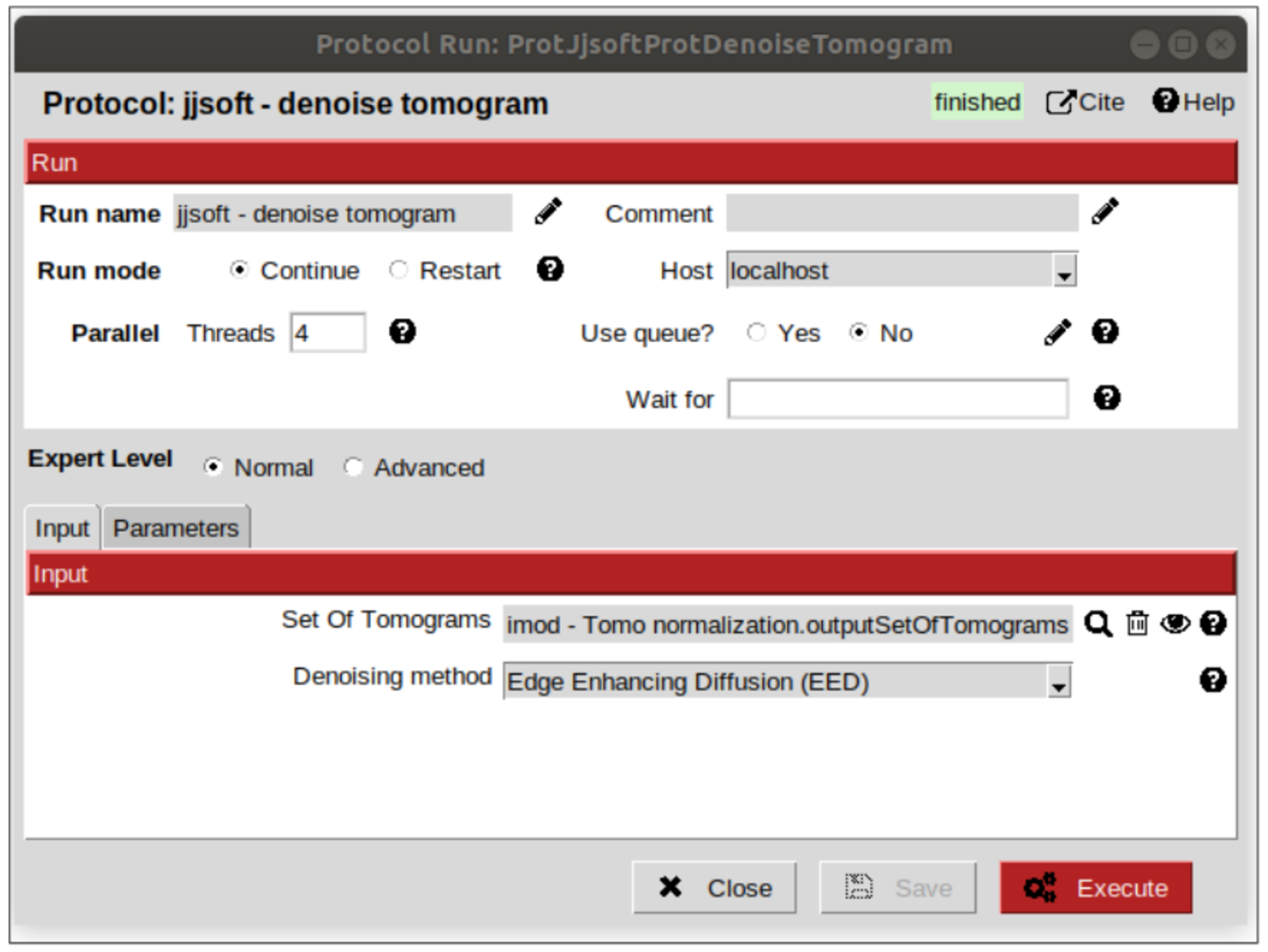
This step is recommended to be carried out before the membrane segmentation, considering that the higher contrast our data has, the better the membranes will be segmented. To do that, open the protocol “denoise tomogram” from plugin scipion-em-jjsoft. Once there, click on the magnifier icon and select, on the pop-up window the pointer to the normalized tomogram (it should be the first on the list, because the objects generated are sorted from newest to oldest by default). Leave the rest of parameters with the default values and click execute the protocol.
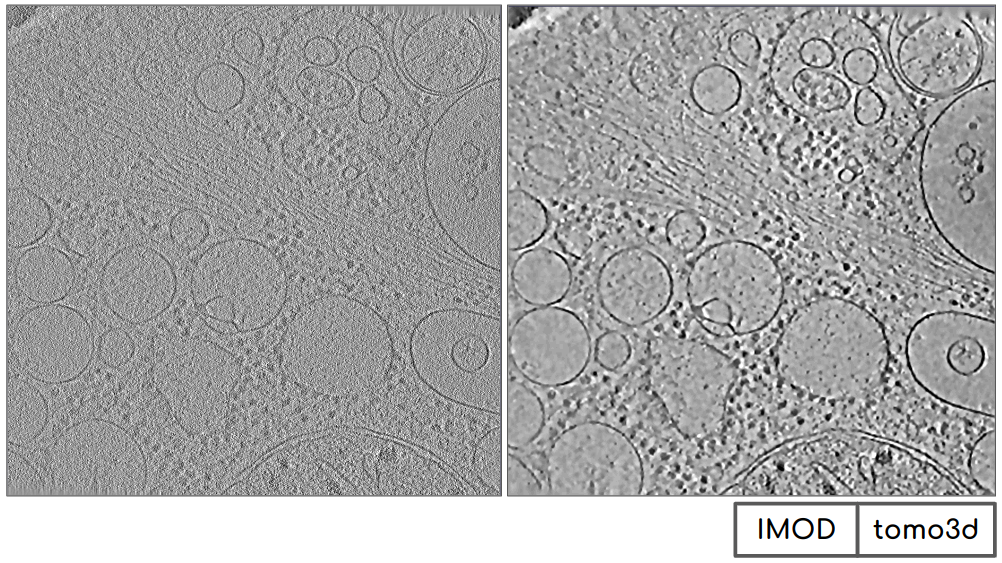
The denoised tomogram can be displayed proceeding the same as explain in section Importing the Tomogram. It can be observed how the contrast has been considerably increased, being the figure on the left the tomogram before the denoising and the one on the right after the denoising.
Membrane segmentation¶
Membrane segmentation and annotation constitute the pre-processing steps for the membrane particles picking with PySeg. The first step will be carried out with protocol “tomogram segmentation” from plugin scipion-em-tomosegmemtv. Open the protocol mentioned and follow the steps listed and illustrated below:
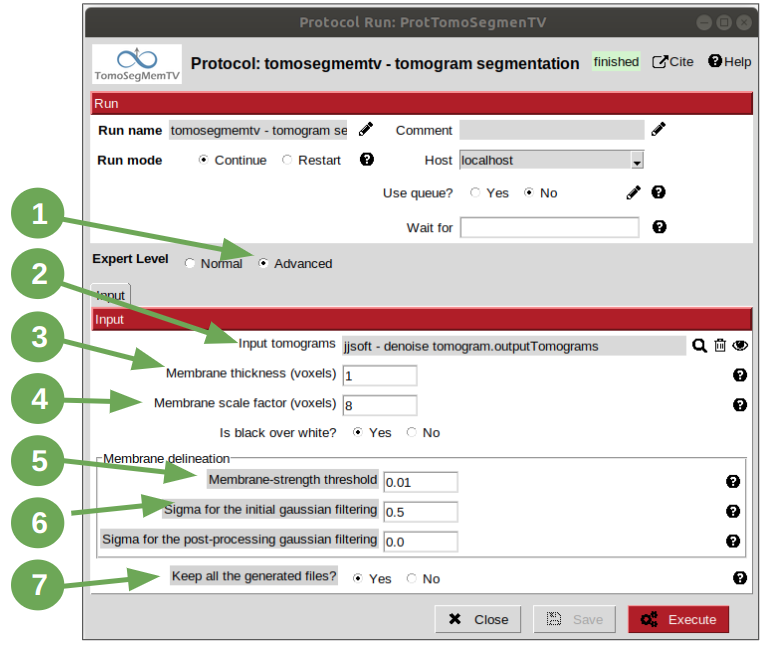
1. Click on Advanced radio button. This action is present in all the protocols that offer advanced parameters and its used to show them.
- Select the denoised tomogram pointer in field “Input tomograms”.
3. Set the “Membrane thickness” parameter to 1 voxel. This is a good and recommended strategy to get the membranes closer to an over-detection scenario than the opposite, which would be the resulting scenario with higher values. In our case, this is the best way to proceed, due to the fact that we’re going to annotate the membranes in the next step with the Membrane Annotator tool, which provides residual structures cleaning tools. Hence, with a low value of this parameter, we’ll obtain less discontinuities in the membranes, but more false positives. The first condition takes to a simpler annotation step in one or two steps per vesicle instead of having to annotate part by part in case of many discontinuities. On the other side, the false positives can be easily removed with the annotation tool.
4. Set the parameter “Membrane scale factor” to 8 voxels. This parameter is used to define the effective neighbourhood of the membranes considered in the calculations (voting process). Hence, this value is recommended to be low for thin membranes and high for thick membranes, and considering the sampling rate of the tomograms whose vesicles are going to be segmented.
5. Set the parameter “Membrane strength threshold” to 0.01. This parameter is used to tune the amount of output membrane points and remove false positives. Lower values will provide more membrane points, at the risk of generating false positives. Thus, this is a critical value when an annotation step is going to be carried out, because a very low value will make most of the structures found in the tomogram to be connected, so it won’t be possible to annotate them separately. On the other hand, higher values will provide a higher probability of the structures to be disconnected, but if the value is too high more discontinuities may be present in the structures detected.
6. Set the parameter “Sigma for the initial gaussian processing” to 0.5. The input tomogram is subjected to an initial Gaussian filtering aiming at reducing the noise so as to determine the derivatives more robustly. By default, a standard deviation of 1.0 voxel is considered. If the membranes are very thin or are very close to each other, use lower values (e.g. 0.5).
7. Set the parameter “Keep all the generated files” to Yes to save all the intermediate results obtained in the different steps carried out internally by tomosegmemTV.
Note: in this example all the parameter values provided have been tuned previously, but in the normal scenario consists of some executions until getting the desired result. Even more, sometimes it is necessary to go back from the membrane annotator to tune some parameter to, for example, get the membranes less connected. On the other hand, it is recommended to keep all the files when you are not familiarized with the algorithm so, if the membranes get lost in the final result, the intermediate results can be analyzed to determine when they got lost and, as a consequence, know know which parameter should be tuned. For a more detailed explanation, click HERE –> AÑADIR REFERENCIA A TEORÍA DE TOMOSEGMEMTV.
The result obtained should look like the figure below.

Hint: the recommended procedure is to work with one or two tomograms of the set to tune the parameters and then use that configuration with all the set.
Membrane annotation¶
Once the membranes have been successfully segmented, they need to be annotated, which means to manually add a numerical label to each to indicate the software that they represent different entities. This step will be carried out with the protocol “annotate segmented membranes” from plugin scipion-em-tomosegmemTV. This is an interactive protocol which generate an auxiliary window that lists the tomograms to be annotated and allow the user to execute the membrane annotator tool by double clicking on it. It also indicates which of them have been annotated and which are still pending to be processed. The only parameter present in this protocol is the pointer to the tomomasks (segmentations).
Note: It may take a few seconds to be displayed after double clicking on one tomogram from the list shown in the auxiliary window.
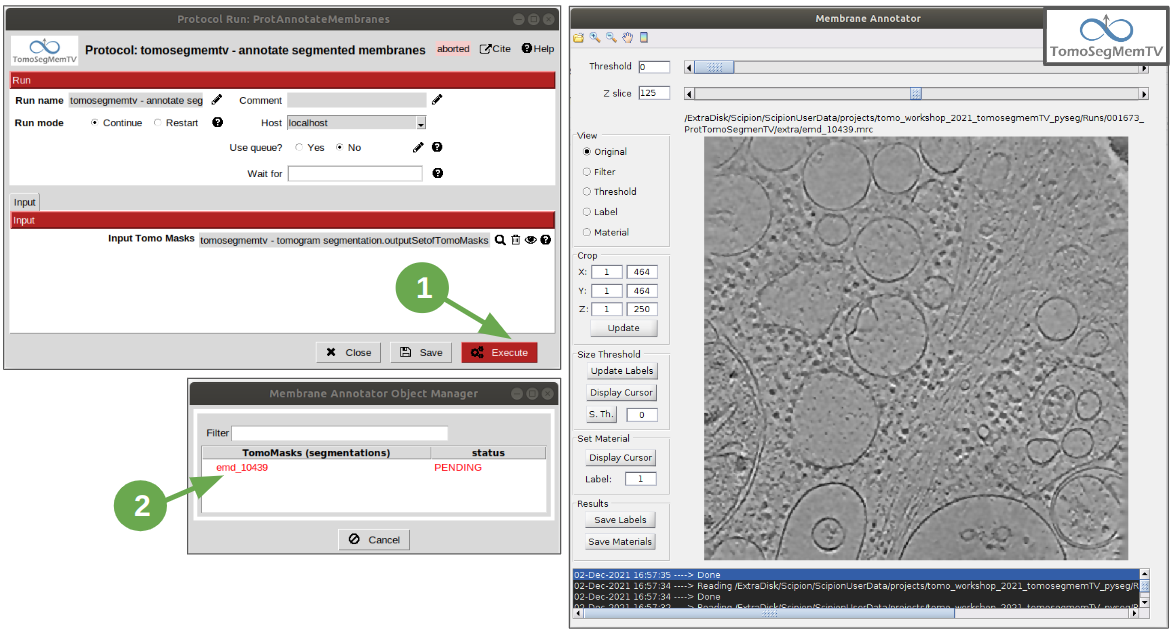
Membrane Annotator overview¶
The following subsections will describe how to use the membrane annotation tool. But before that, let’s have a quick look at its interface and components:
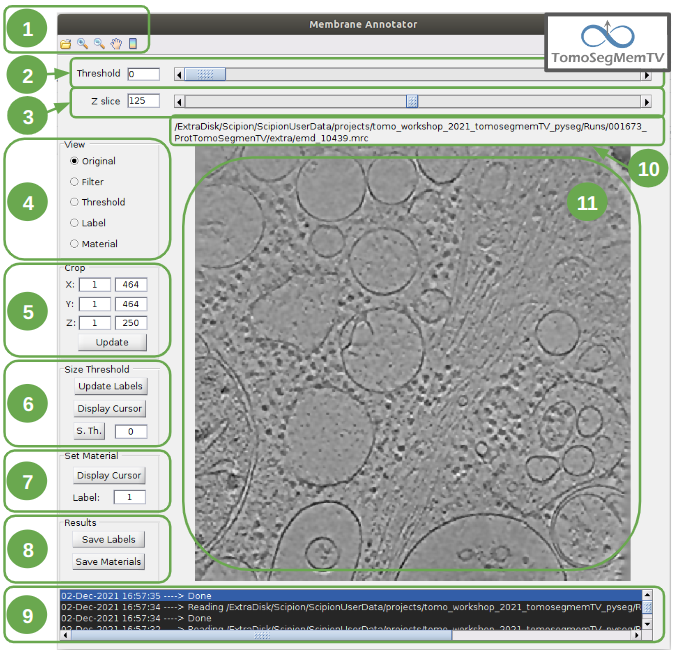
Here is a brief explanation of each of the component enumerated in the figure above:
1. Tools shortcuts: it offers useful functionalities to work with the structures found in the loaded tomogram, like the zoom in/out or the click and drag.
2. Density thresholding tools: the thresholding is the starting point of every labelling procedure. It’s value can be updated using the slider or introducing a value in the corresponding textbox.
3. Z slice navigation tools: another textbox and another slider are provided to navigate through the Z slices of the tomogram and locate all vesicles desired to be annotated.
View panel: it allows to visualize different representations of the loaded data:
4.1 Original - current tomogram data
4.2 Filter - input of the density thresholding operations.
4.3 Threshold - output of the density thresholding operations.
4.4 Label - Result of “Update Labels” operation (assign to each structure a label which is its size in voxels.
4.5 Material - Result of the manual labelling. It shows the annotated membranes with the assigned value.
Crop panel: it can be used to crop the tomogram oroviding the X, Y and Z ranges and clicking in button “Update”.
Size Threshold panel: it can be used to perform three different operations:
- 6.1 Update Labels: automatic labelling of the structures found depending in the density threshold value. It assigns,
by default, the size of each structure as label. It will update the view to the view “Label”.
- 6.2 Display Cursor: it’s used to check the size of each structure. One click on it will activate the cursor mode,
which will display the value of the pixel selected. To finish this cursor mode, click again on the previous button, whose name will be now “Stop Cursor”. This functionality is very useful to determine if, for example, the different parts of a discontinuous structure have been detected as parts of the same structure of not and manually annotate them coherently.
- 6.3 Size Thresholding: it can be used to remove undesired sizes of structures, like the ones which are too small.
To do that, simply introduce a size value in the textbox and click on the button “S. Th.”.
7. Set Material panel: it works like the “Display Cursor” functionality explained in 6.2, but to annotate the desired structures. To do that, click on button “Display Cursor” to activate the cursor mode. Then select a structure by clicking on it (until here it’s the same as before) and finally introduce a value in the corresponding textbox before clicking again on the cursor button (renamed to “Change Lbl.”) to stop it and automatically execute the labelling of the selected structure, shown in view “Material”.
8. Results panel: it has two buttons, one to save the automatic size labels calculated when clicking on button “Update Labels” and the other to save the manually annotated structures. IMPORTANT: working from Scipion, this step is required to be carried out once all the desired vesicles have been annotated.
- Log panel: it registers the main actions that have been carried out by the user.
- Tomogram file name: informative.
- Data visualization panel.
Target vesicles¶
It can be observed that three of the vesicles (squared in the figure below) contain most of the membrane ribosomes. These are the ones we’re going to annotate.

Density thresholding¶
First of all, let’s set the density threshold value [2] to 0.05. This value offers a clean and continuous view of the different structures present in the loaded tomogram.
Hint: to get an intuition of how the variations in the density threshold value affects the data, it’s very recommendable to test different values until a promising visualization is obtained.
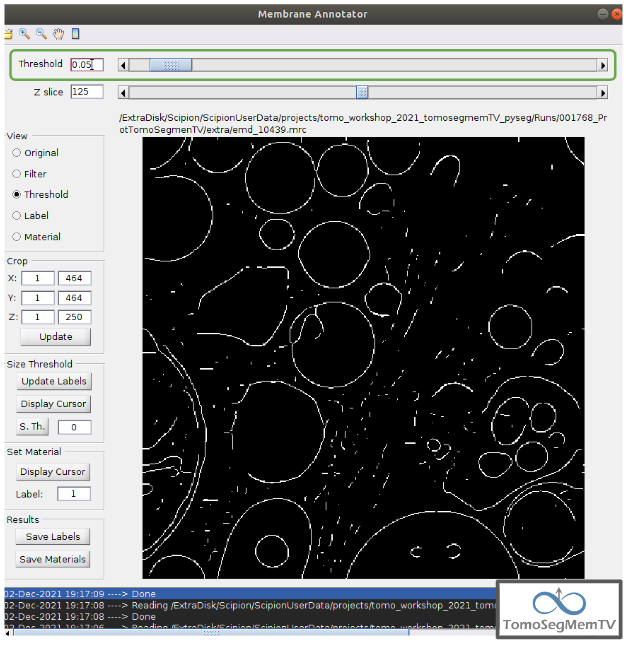
To check the results, click on button “Update Labels” [6]. The result of this operation should look like as the figure below. It can be observed that the segmentation and density thresholding values were correctly determined because all the target structures present different colors, which means different sizes. In some cases, like in target 1, there are two or more different colors (sizes) for the same vesicle, but this is more than normal in the case of our data (in situ tomogram). This can be solved annotating the different parts with the same label.
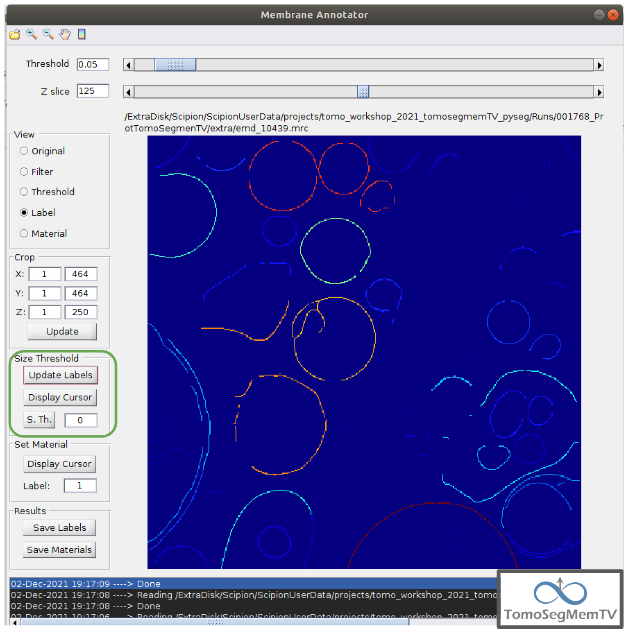
On the other hand, it’s recommendable to check that both parts of target 2 are of the same size. It can be easily done with the button “Display Cursor” from panel “Size Thresholding” [6]. The result is that in this case both parts are of the same size, which means that most of the whole changing shape through the slices was very well segmented.
Manual annotation of the target 1¶
The first target membrane has been detected in two unconnected parts of different sizes (colors), as shown below (the size is shown in the index label of the tooltip. The background size will be always 0). It can be observed that target 3 has different size, so it’s not connected to the orange part of target 1 and that the blue part of target one can be annotated with the same label as the orange one to get the full membrane annotated.
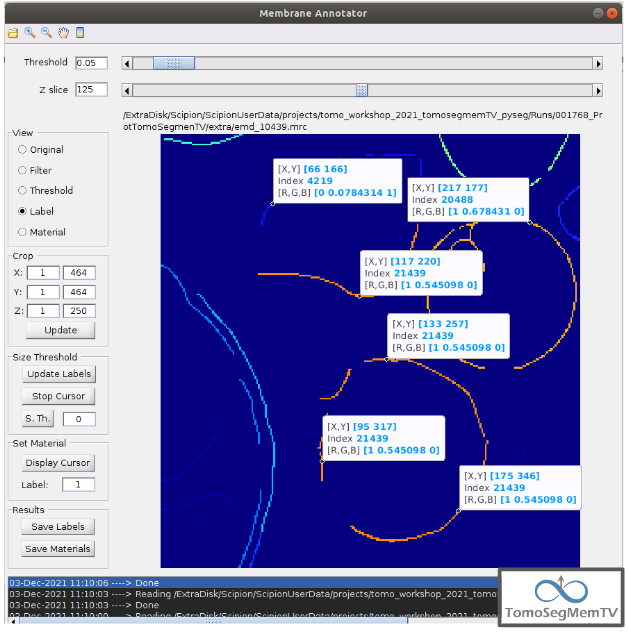
The procedure followed to check the sizes was:
- Click on the magnifier with a cross icon from “Tools shortcuts” [1].
2. Create a zoom window clicking and dragging around the target 1 vesicle to zoom in. When the zoom mode is active, it can be smoothly controlled with the mouse wheel.
3. Click on button “Display Cursor” from panel “Size Threshold” [6] and click on the structure whose size is desired to be displayed. To fine tune the position of the cursor, use the arrow keys from the keyboard. Note: to generate multiple tooltips, right click on the current tooltip and select option “Create New Data Tip” or directly press shift + left click.
- To finish the cursor mode, click on the same button pressed to activate it, but now called “Stop Cursor”.
Let’s annotate now the orange part of target one with label 1 (Use the zoom in tool if necessary, as explained before):
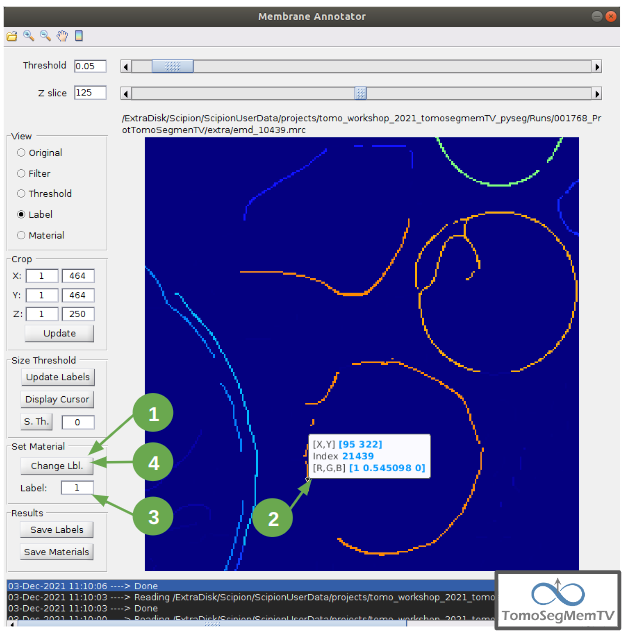
- Click on button “Display Cursor” from panel “Set Material” [7].
2. Click on the membrane and, before clicking on the same button (now named “Change Lbl.”), be sure that the clicked pixel belongs to a structure (index must be grater than 0).
3. Leave the textbox “Label” value as 1. If we we annotating the target 2 o target 3 vesicles, this value should have to be set to 2 or 3, respectively.
4. Finally, click on the button “Change Lbl.” to annotate that part of target 1 vesicle with label 1. This action will display automatically the view “Material” from the panel “View” [4], as can be observed in the figure below.
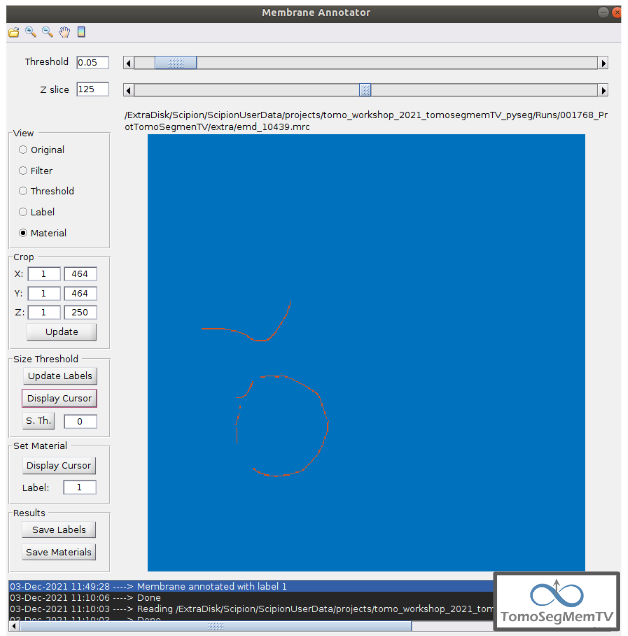
If we repeat this procedure with the blue part of target 1 vesicle (annotatin it with label 1), the result should look like as shown in the figure below.
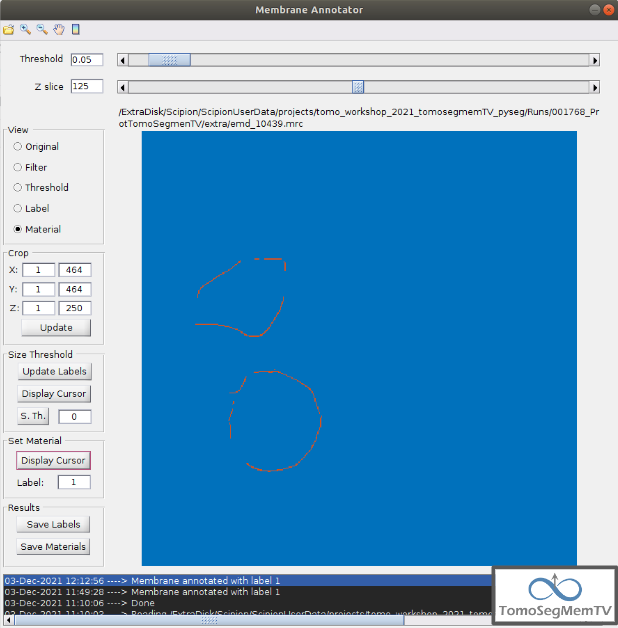
Manual annotation of the target 2¶
Proceeding the same as explain in section Manual annotation of the target 1, it can be observed that the target has been detected in two different parts (upper part, with a size of 111171 voxels and lower part, of size 10330 voxels), just the same as what happened with target 1. Moreover, the inner small vesicle and the top left structure are disconnected from target 2, because they have different sizes (see figure below).
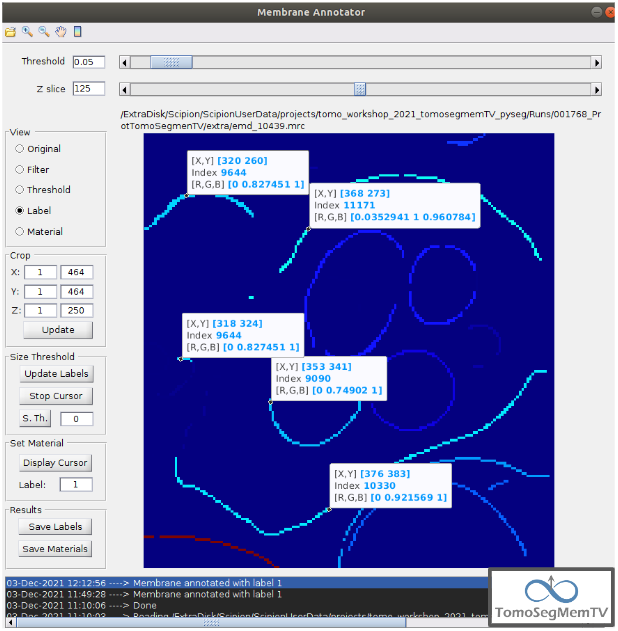
Hence, we can proceed to the manual annotation, this time with label 2. The final result of the target 2 vesicle annotation is shown in the figure below.
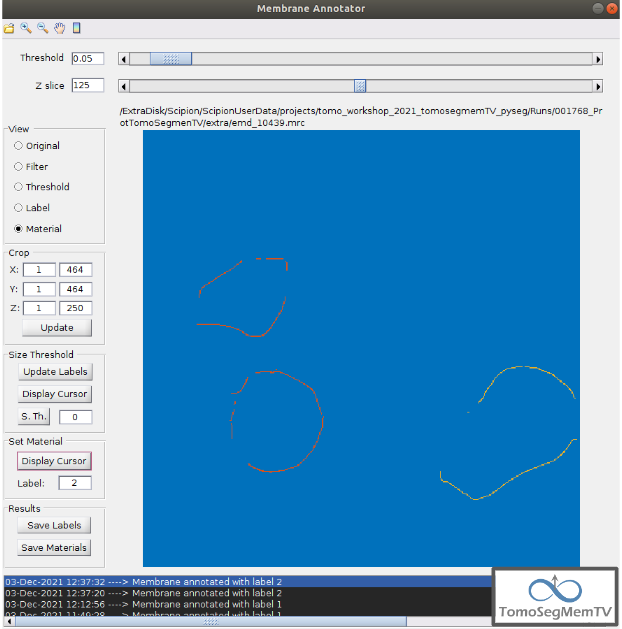
Manual annotation of the target 3¶
This is the easiest one, identified as a continuous structure. So we can directly annotate it with label 3. The result of the three membranes annotated can be observed in the figure below.
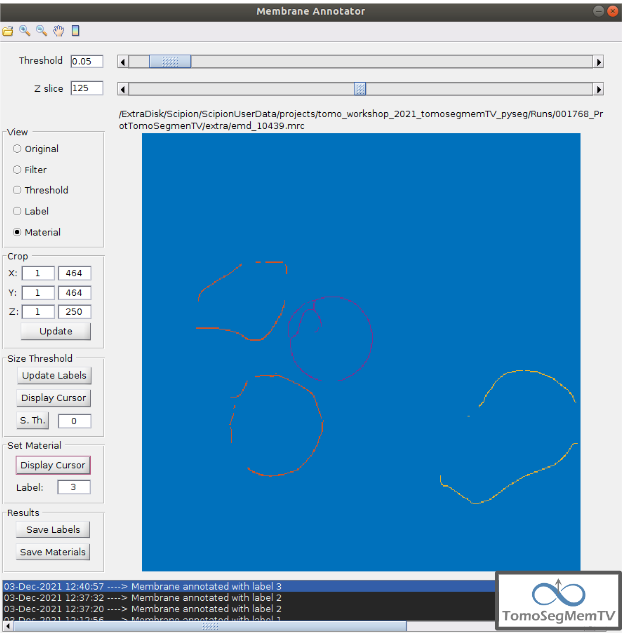
Save the annotated vesicles and finish the interactive annotation protocol¶
To successfully save the results of the annotation, follow the steps enumerated below:
- Click on button “Save Materials” from panel Results [8].
- If everything goes fine, the first line of the “Log Panel” [9], should be “Materials were correctly saved”.
3. Close Membrane Annotator and check that the status of the tomogram listed in the auxiliary window has been updated to “DONE”. Finally, close the auxiliary window.
4. The protocol box should have now update its state to inactive. If not, refresh the project interface (refresh icon is located at the top right corner of the project panel).

Analyze the annotated membranes¶
If we click on button “Analyze Results” in the lower panel of the project interface, the 3D visualization tool from plugin scipion-em-tomo3d is launched. It allows the user to observe the membranes annotated placed on the full tomogram or by slices, as shown in the figure below.

Resize the tomomasks (segmentations)¶
After having carried out the segmentation and annotation of the vesicles in a smaller size to improve both performance and contrast (explained in section Tomogram normalization), the segmented and annotated data must be resied to its previous size for the picking of the membrane particles (smaller sampling rate will make the picking algorithms easier and even possible to find the desired densities). This operation will be carried out with protocol “Resize segmented or annotated volume” from plugin scipion-em-tomosegmemtv. The tomomasks desired to be resized and the tomograms to which they have to be referred and resized to their size are the arguments required to be filled. Select the pointer to the annotation protocol output for the first and the pointer to the imported tomogram for the second.
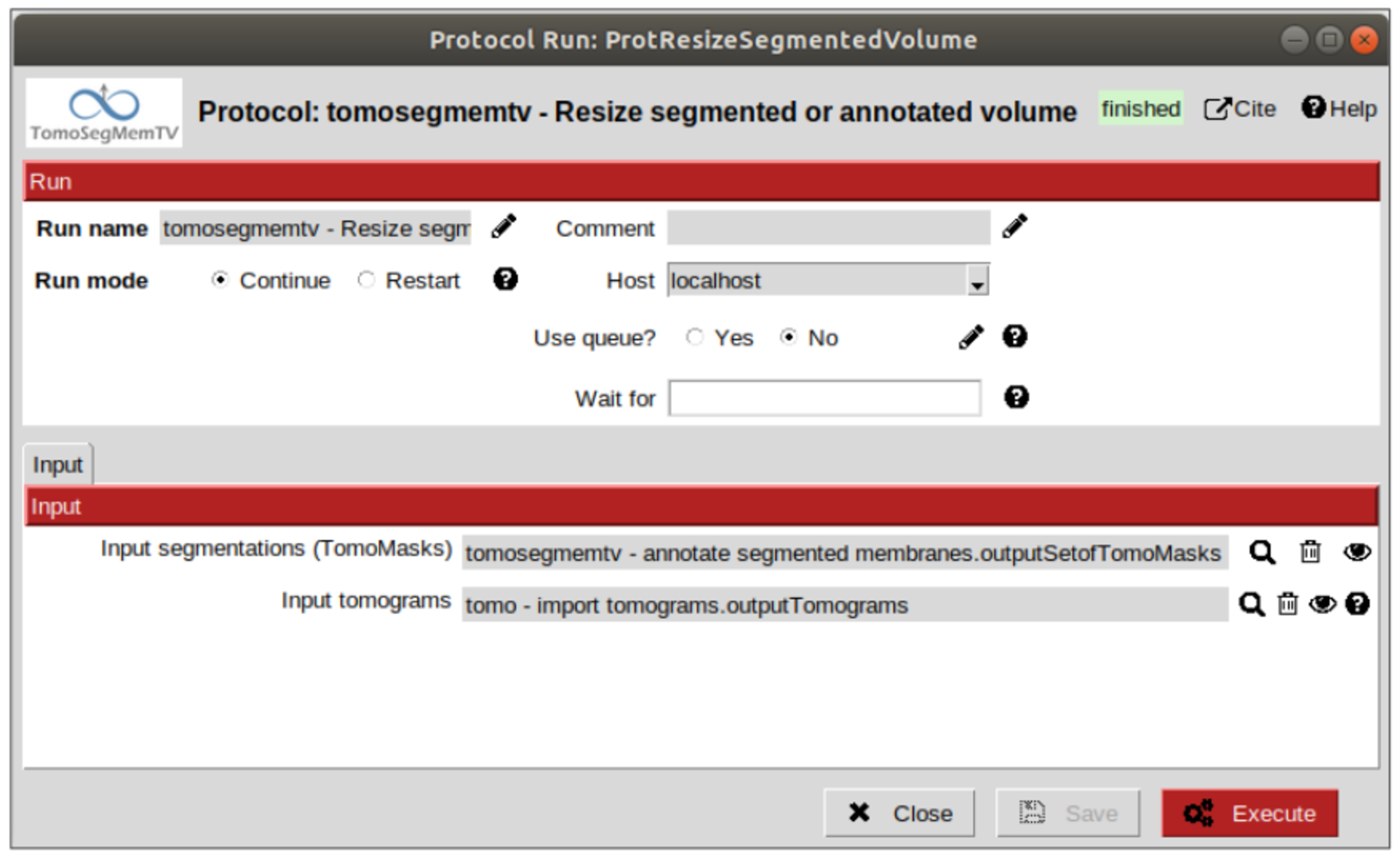
We’re referring the tomomasks to the imported tomograms and not to the denoised ones to carry out the picking procedure with the less processed data as possible, for two main reasons:
1. PySeg graphs calculations expect the data not to be filtered, so it will provide the best result with unfiltered (e. g. not denoised) data.
2. Avoid all the interpolations and mathematical treatment of the data at the pint of identifying small structures, increasing the probabilities of the picked objects to be a physical entity instead of a mathematical artifact,
SUMMARY:
At this point we have the membranes segmented, annotated, at the correct size and referred to the imported tomograms. Thus, we’re ready for the picking.
Directional picking with PySeg¶
As it was explained in PySeg presentation, the directional picking is composed by four main steps (assuming that the segmentation and annotation of the membranes have been performed before):
- Preseg: segment membranes into membranes, inner surroundings and outer surroundings
- Graphs: analyze a GraphMCF (Mean Cumulative Function) from a segmented membrane. A graph is a set of connected nodes.
3. Fils: filter a MbGraphMCF object by extracting a filament network. A filament represent to nodes connected (only the first and last nodes, without intermediate elements).
- Picking: extract particles from a filament network of a oriented single membrane graph.
Each of these steps is represented with a different protocol inside Scipion, and they will be explained in the following subsections.
Preseg¶
Look for pyseg protocol and open it. At first sight, it’s remarkable that this protocol allows the user to get the previous segmented and annotated data from Scipion (Scipion Protocol) or from outside (e. g., using the standalone version of the membrane annotation tool and preparing a star file with the data as expected by the preseg.) Said that, let’s replace the following parameter default values by the ones required for this tutorial:
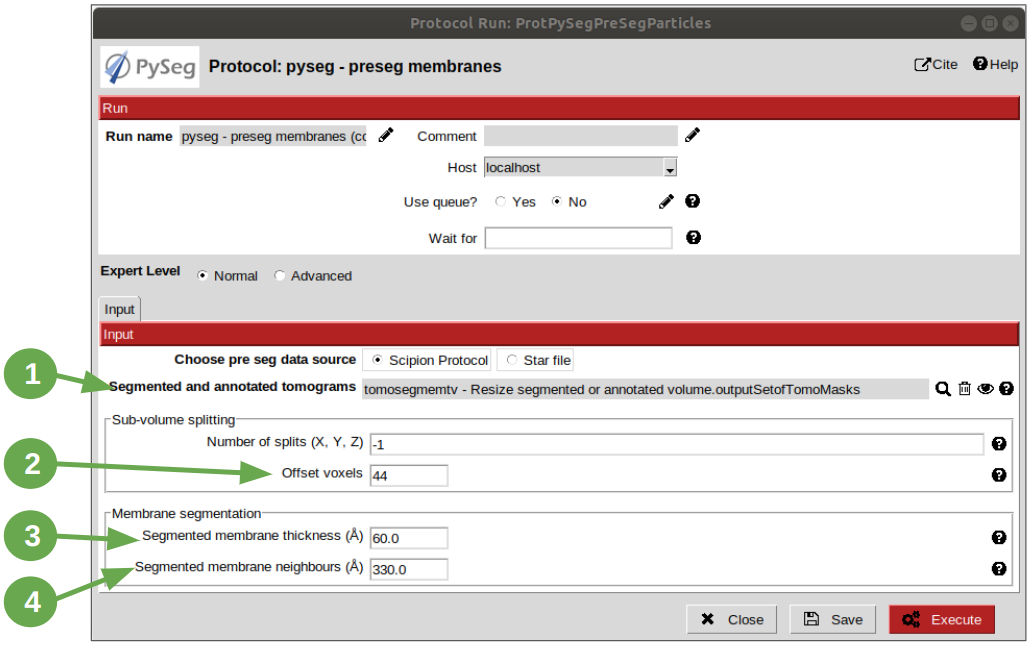
1. On parameter “Segmented and annotated tomograms”, select the pointer which corresponds to the output of the resizing protocol applied before.
2. Update value of parameter “Offset volxels” to 44 voxels. This parameter represents the width of a margin considered when cropping the vesicles. It’s necessary to provide a value which ensures that the desired biological entities, e. g. membrane proteins, are included in the cropped area.
3. Update “Segmented membrane thickness” to 60 angstroms. Value introduced will be divided by 2 internally to get the semi-width of the membrane, which which will be considered at both sides of the membrane central line.
4. On parameter “Segmented membrane neighbours”, type value 330 angstroms. This parameter represents the thickness around the membrane to represent the in-membrane and out-membrane surroundings desired to be included in the analysis. The value chose was 330 angstroms because the size of a ribosome varies from 200 to 300 angstroms in diameter, and a margin of the 10% of error is considered for the biggest size (that additional 30 angstroms).
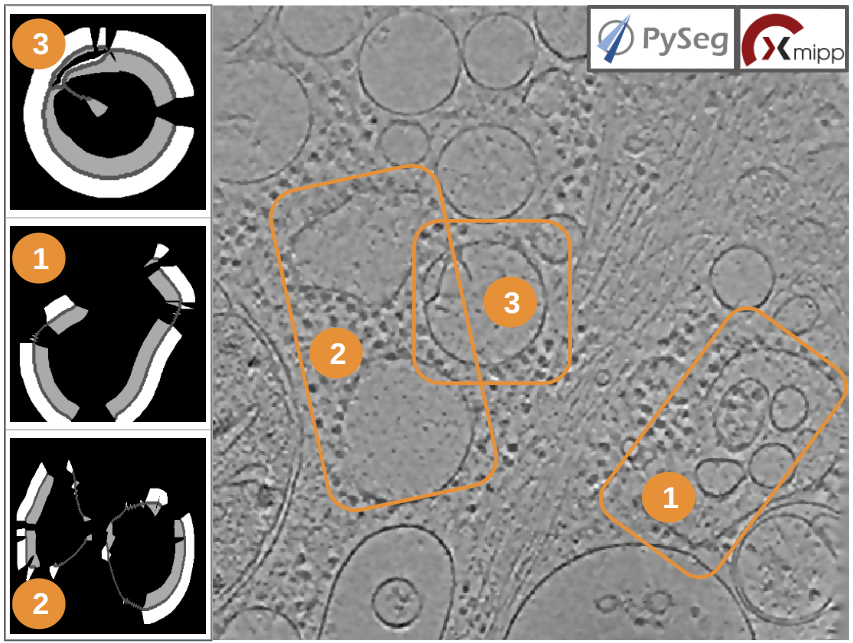
If the results are displayed with the viewer DataViewer from xmipp (right click in the output element shown in the object lower panel, in tab “Summary”.), they should look like as can be observed in the left side of the figure below, which represents the area segmentation of the central slice of each vesicle. The right side and the numbers are used to visually relate each segmentation to the target vesicles they represent.
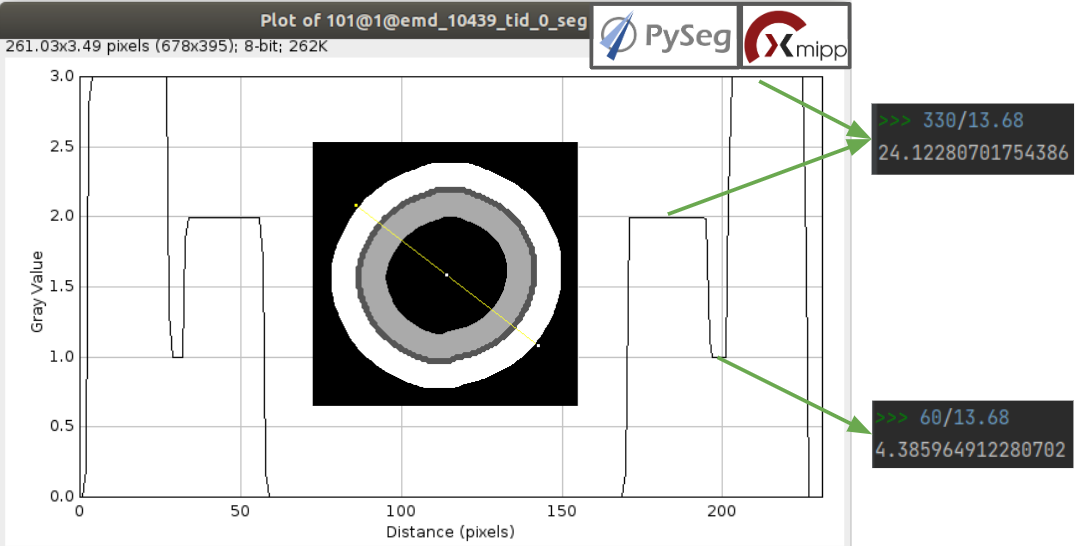
For a better understanding of the parameters introduced in this protocol, the figure below shows the thickness of the membrane, the inner surroundings and the outer surroundings and their conversion to angstroms considering the sampling rate, which is 13.68 Å/voxel. The graph shown is the result of tracing a profile on one of the slices of target vesicle 3. This was done also inside Scipion, using the tools included in the viewer DataViewer from xmipp.
Graphs¶
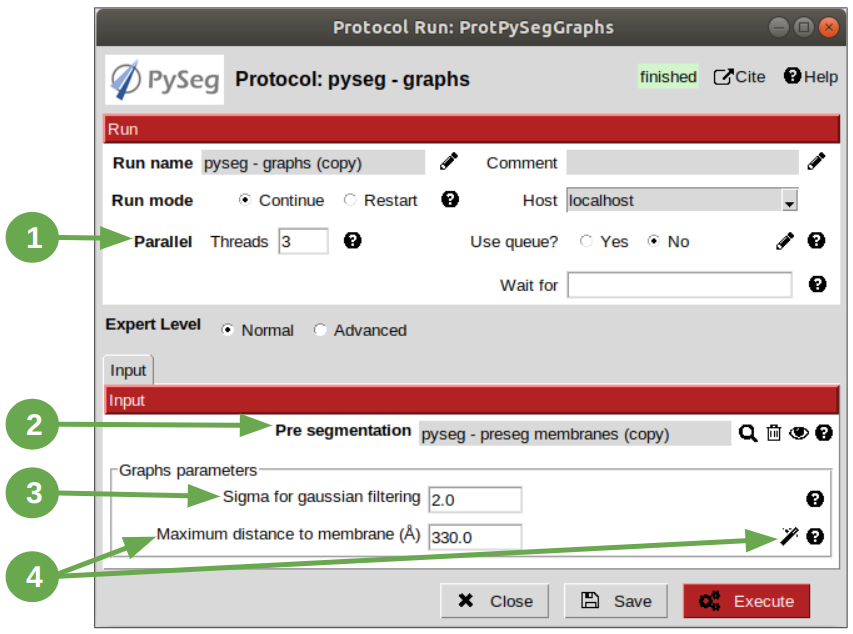
At this point, it’s time to calculate the graphs: look for the protocol, open it and update the parameter values as enumerated below:
- Set parameter “Threads” to 3.
- Set parameter “Pre-segmentation” pointer to the preseg protocol executed before.
3. Update parameter “Sigma for gaussian filtering” to 2. It allows to smooth small and irrelevant features and increases the signal noise ratio (SNR). Higher values will provide less dense graphs (lower execution time), so they should be used when picking large particles, like ribosomes.
4. Parameter “Maximum distance to membrane” can be set in two different ways, which are introducing manually the desired value or clicking on the wizard (wand) icon. This action will read the value of parameter parameter “Segmented membrane neighbours” from the preseg protocol selected in parameter “Pre-segmentation”. That value should be 330 angstroms.
Results can be displayed by clicking on button “Analyze Results”. That action will allow us to select which vesicle is desired to be represented with 3D viewer from plugin scipion-em-tomo3d setting the coloring option “Color Graph By”, located on the top left corner, to value “mb_eu_dst”, which colors the graphs considering the euclidean distance to the membrane. Results should look like shown in the figure below. Observe that the numbers correspond to the target vesicles which is being used in this tutorial from the annotation step.

Fils¶
Once the graphs have been calculated, it’s time to refine them. This is the aim of the fils protocol. This is a good moment to go back to the PySeg presentation and refresh the concepts of euclidean and geodesic distances and sinuosity. Apart from that, the protocol labels were written with the objetive of providing an approximate idea of what these concepts means.
Now, let’s open the fils protocol and set the following parameters as explained below:
- Input tab: set the parameter “Graphs” pointer to the graphs protocol executed before.
2. Sources tab: used to define geometrically how the filaments should be in the area selected as source area. Observe that the source filament area is the membrane. Because the ribosomes doesn’t go through the membrane, the geometrical descriptors on this area won’t make a difference in the obtained result. Hence, let all the parameter with the default values. Targets tab: it’s the same as the sources tab, but for the area chosen as target area:
2.1 Set the parameter “Filament area” to “Outer Surroundings”. This is the area of interest for picking the membrane ribosomes.
2.2 For the euclidean distance, set the minimum value to 0 nm and the maximum to 30 nm, which is the largest size expected for the ribosomes we’re trying to pick.
2.3 For the geodesic distance, set the minimum value to 0 nm and the maximum value to 60 nm. That way, we’re considering some flexibility in the filaments.
2.4 For the sinuosity, set the minimum value to 0 and the maximum to 2. The recommended value for this parameter is the ratio geodesicLength/euclideanLength, but it doesn’t have to. Sinuosity specified in a value of distances or lengths contained in the intervals set before for euclidean distance and geodesic length, respectively.
3. Refinement tab: it’s used to apply a geometric filter to refine the calculated filaments. They must be introduced in ranges [min max]. In our case, considering the type and and features of the target particles, set them as follows:
3.1 Euclidean distance range: from 20 to 30 nm, which is the expected range of a ribosome size variation, approximately.
3.2 Geodesic distance range: from 20 to 60 nm, which goes from the shortest straight length to a maximum value considering some flexibility.
3.3 Sinuosity range: from 0 to 2. Thus, we’re considering all the flexibility values present considering the euclidean and geodesic values provided before.
Note: the lengths are delimited by the thickness of each area generated in the preseg protocol.
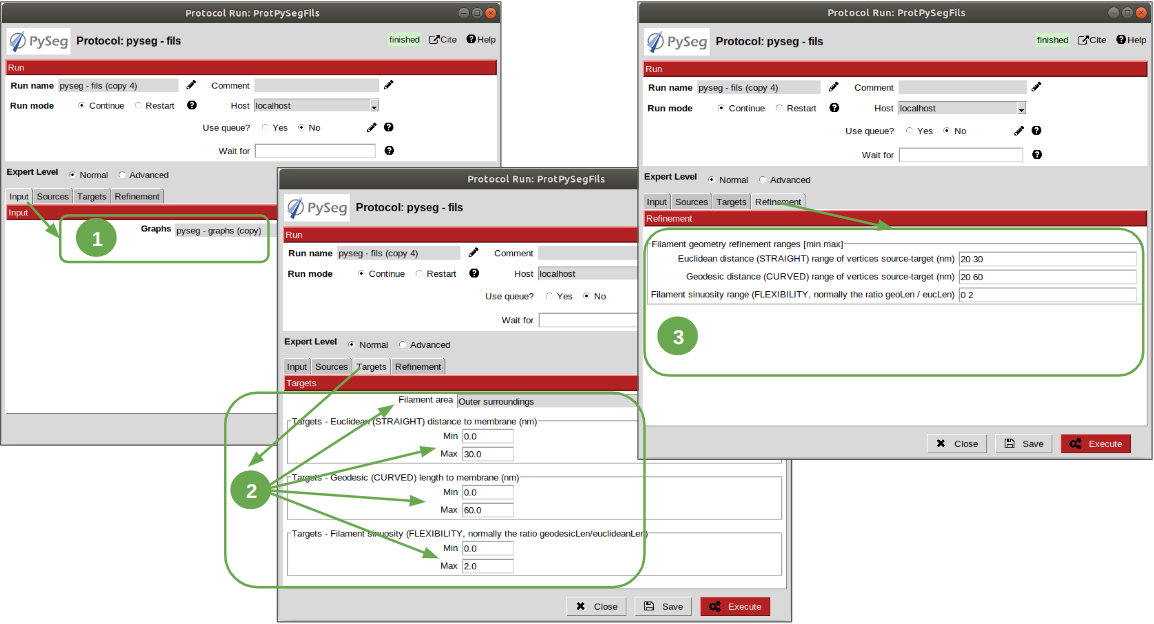
The resulting filaments should look like in the figure below. The same considerations as in the graphs protocol results have been followed.

Picking¶
Finally, to get the particles picked, let’s open the picking protocol and set the following parameters as follows:
1. Input tab: we have to select which filaments protocols to use and which set of tomograms must be the coordinates referred to. In our case, we only have the previous fils protocol execution, and the coordinates should be picked on the original tomogram, following the same as raw data as possible reasoning as before to avoid possible mathematical artifacts.
Picking tab:
2.1 Set the parameter “Segmentation area for picking” to “Outer surroundings”, where the ribosomes are located.
2.2 Set parameter “Find on two surfaces” to “Projected local minima”. This parameter is used to indicate if we want to keep the coordinates of the cutting point of the filament with the membrane or the cutting point and the projections of the filament over the membrane, respectively. The second option will result in an over-picking. This can be a good strategy in order to ensure that no particles are lost when picking, but some kind of distance or angular filtering should be applied later to remove the duplicates.
3. Refinement tab: this tab allows the user to refine the picking results by specifying the density level or the minimum distance between the picked coordinates. Let this tab with the default values. We’ll deal with the over-picking later.
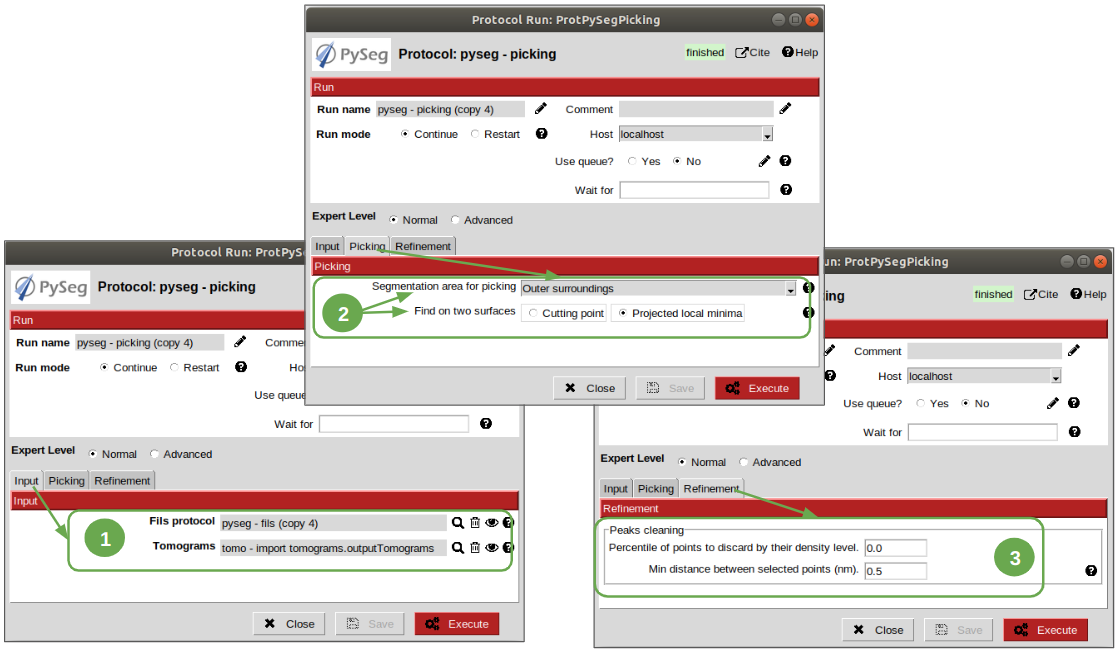
It can be observed in the summary tab of the lower panel on the project interface that 2339 particles were picked. For the moment, let’s ignore the box size displayed there, which is a default value required for some viewers to be different from zero.
Results can be displayed with multiple viewers, like the one from plugin scipion-em-emantomo but, following the same structure considered to show the results on the graphs protocol and fils protocol, we’ll use the viewer from plugin scipion-em-tomo3d:

Picking post processing¶
This section contains the steps suggested to resolve the over-picking scenario described in picking protocol and also to get rid of bad picked elements. For the first one, we’ll use the protocol “remove duplicates” and for the second, the protocol “filter by normal”, btoh from plugin scipion-em-tomo3d.
Remove duplicates¶
Using this protocol, the over-picked particles will be replaced by the mean position and orientation of them. Hence, let’s open the protocol, select the pointer to the coordinates picked before and let the radius value with the default value of 10 voxels. This is only a coincidence, considering half of the size of the biggest ribosome and the sampling rate of our data (150Å / 13.60 Å/voxel ~ 11 voxel).
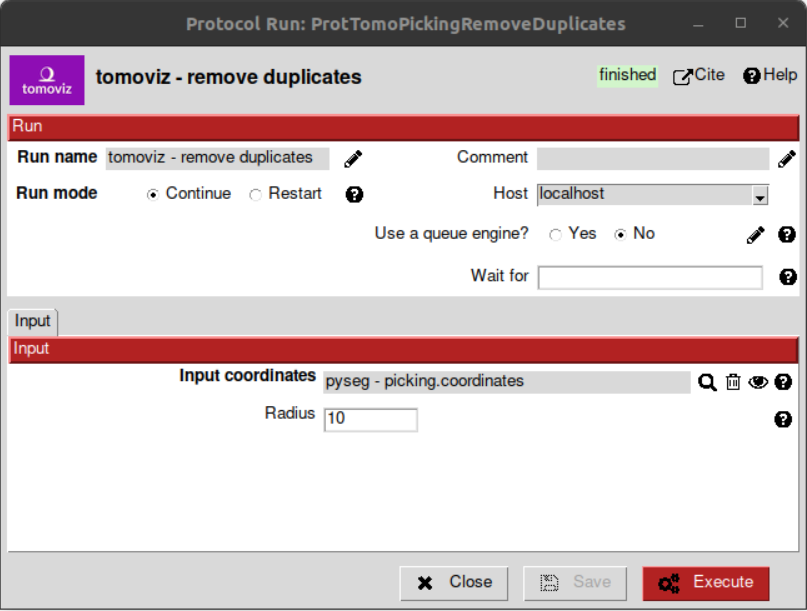
Again, on the summary tab of the lower panel on the project interface, it can be observed that we have now 641 particles after having removed the duplicates. As before, using the viewer from plugin scipion-em-protocol, the result should look like this:

Filter by normal¶
Let’s continue cleaning the data. Protocol “filter by normal” takes the vesicles and the particles and filters them by different criteria related with the normal direction. If the user has a set of coordinates with orientation but not the surfaces or meshes corresponding to the membranes or vesicles which are the reference for the orientation, these surfaces can be created from the orientated coordinates by using the protocol “fit vesicles” from plugin scipion-em-xmipptomo plugin. Hence, let’s generate the meshes required to use to use the filter by normal.
Protocol “fit vesicles” only requires two inputs, which are the pointers to the resulting set of coordinates after having removed the duplicates and the tomograms from which the input coordinates come. Finally, click on button “Execute” and the set of meshes will be generated.
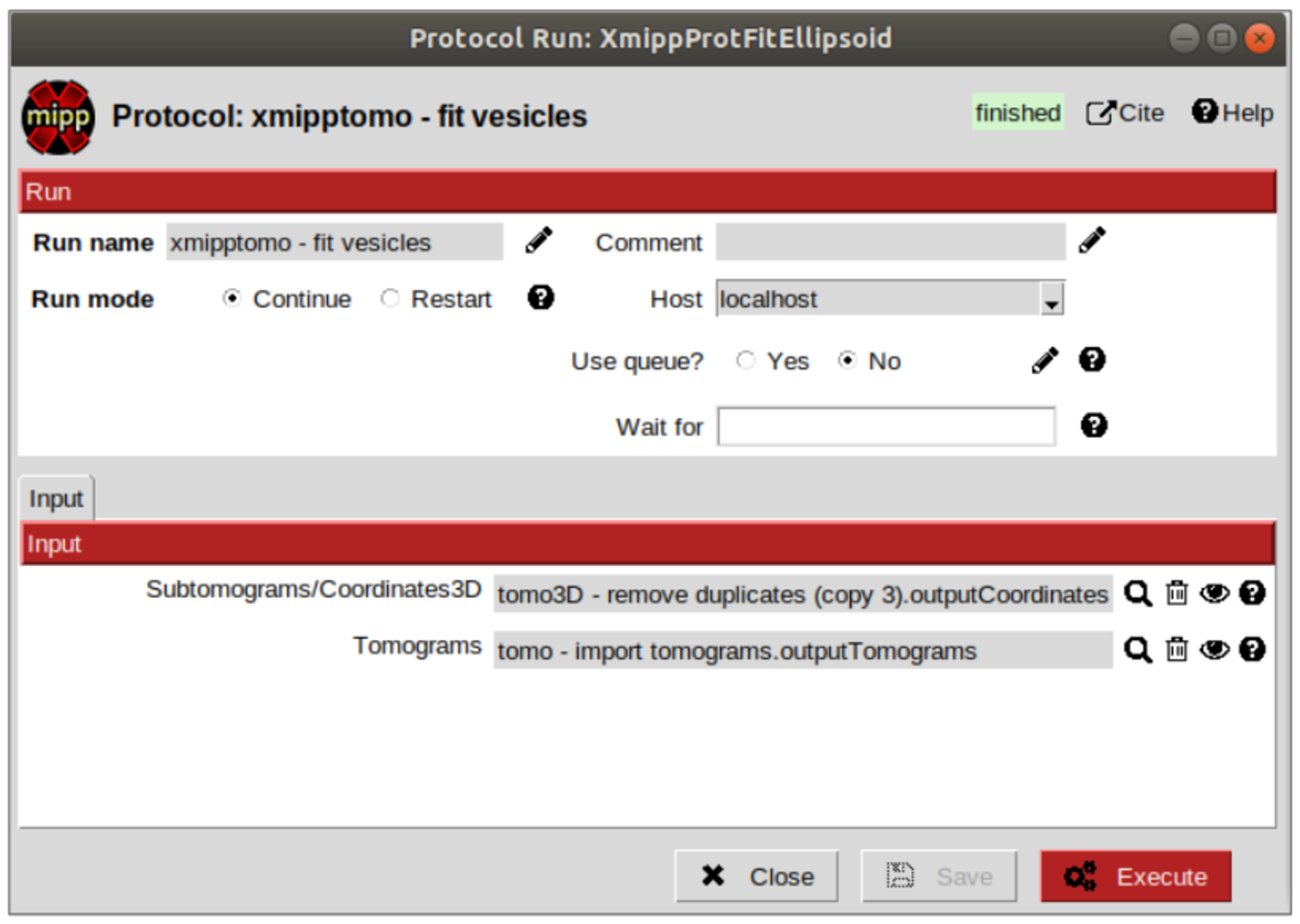
At this point, we are ready to use the filter by normal, so let’s open it and follow these steps:
- Set the input coordinates pointer to the coordinates obtained after having removed the duplicates.
- Set the vesicles pointer to the set of meshes generated before with the protocol “fit vesicles”.
- Update the parameter “Tolerance in degrees” to “30”.
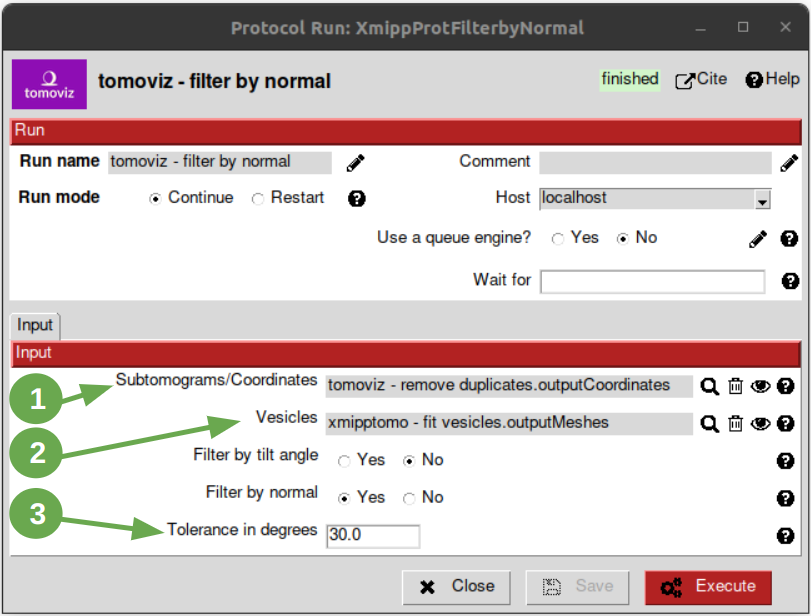
After executing it, we should have 285 items.
Subtomogram extraction¶
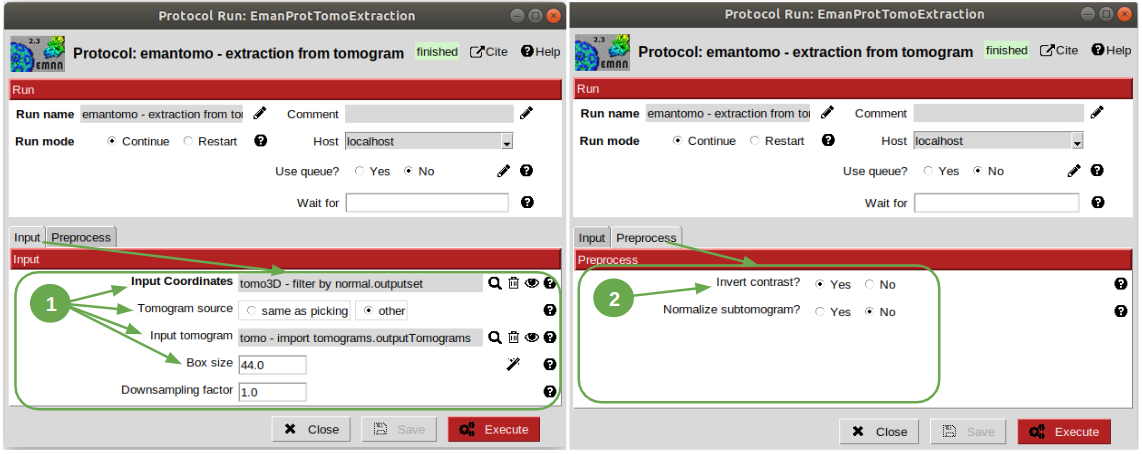
This operation consists on cropping out particles with a specified box size in order to get them separately and with the surroundings to perform the subtomogram averaging. We’ll carry it out using the protocol “extraction from tomogram” from plugin scipion-em-emantomo. Let’s open it and set the parameters as listed below:
Input tab:
1.1 Set the input coordinates pointer to the coordinates generated after having filtered by normal.
1.2 Set the parameter “Tomogram source” to “other” to manually specify the tomogram from where the particles were picked.
1.3 Set the pointer of the input tomograms to the imported tomograms (remember, as raw data as possible).
1.4 The box size is quite critical. Let’s ignore the wizard considering that PySeg considers the coordinates from the membrane, so the box size introduced to ensure that the whole particle is contained in the cropped subvolume must be approximately the double of the particle largest expected size, which is 300 Å. Thus, in voxels it should be around 600Å / 13.68Å/voxel ~ 44 voxel.
Preprocess tab:
2.1 Set tha parameter “Invert contrast” to “Yes” to get, on our case a white over black representation.
2D classification¶
With the particles extracted, we’re almost ready to carry out a 2D classification with a protocol of the same name from plugin scipion-em-pyseg. This 2D classification is performed using a clustering algorithm of the rotational average of each particle around the normal axis. But, before that, let’s deal with that ‘almost ready’. We’re not ready yet because the classification protocol needs a mask which is applied to work on the regions of interest of the subtomograms. In our case, the region of interest is the membrane and the ribosome.
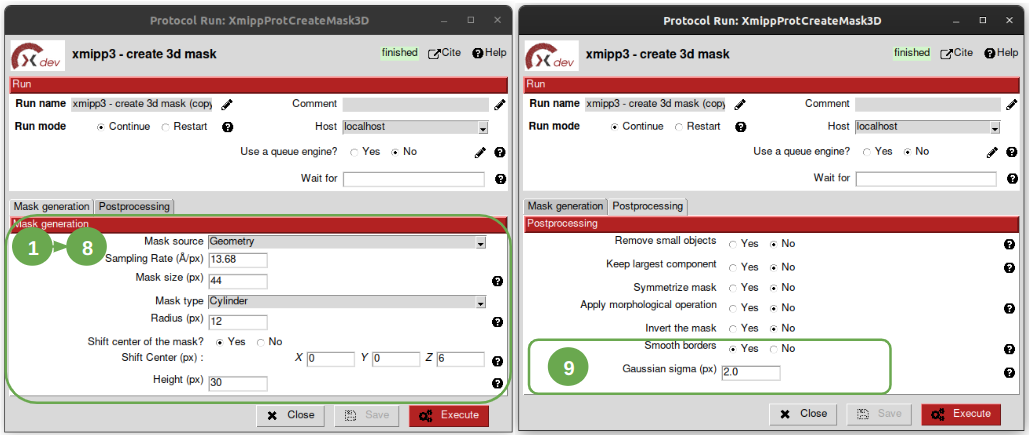
We’ll generate the mask with protocol “create 3d mask” from plugin scipion-em-xmipp. Our mask will be a cylinder of radius approximately half of the size of the biggest ribosome considered and with a height enough to cover the whole ribosome, the membrane and a small amount of the inner surroundings. All these requirements together and the fact that the mask will be referred to the center of the box (which means the vesicle), also suggest the need of some shifting of the cylinder center.
Now, let’s open the protocol and set the following values in the parameters listed below:
- Set the parameter “Mask source” to “Geometry”.
- Set “Sampling rate” to 13.68 Å/px to make the mask be at the same sampling rate of our data.
3. Set “Mask size” to 44 px, because it has to be of the same box size as our subtomograms (44 is the value we introduced as box size ehrn extracting the particles with scipion-em-emantomo).
- Select “Cylinder” from “Mask type”.
5. Set “Radius” to 150Å / 13.68Å/px ~ 11 (+ 1 leaving some margin to ensure the particle is completely contained in the mask). Thus, the radius will be set to 12 px.
- Set the parameter “Shift center of the mask” to “Yes”.
7. Set the Z component of the parameter “Shit Center” to 6 px, which is about 6px * 13.68Å/px ~ 82Å, which is approximately 60Å (remember preseg protocol) of the membrane thickness and 20Å of the inner surroundings.
8. Set the parameter “Height” to 30 px. This value was estimated as 300Å (ribosome largest size) + 60Å (membrane thickness considered) + 20Å (inner surrounding considered), which is 380Å / 13.68Å/px ~ 28px which will be considered 30 to leave a small margin.
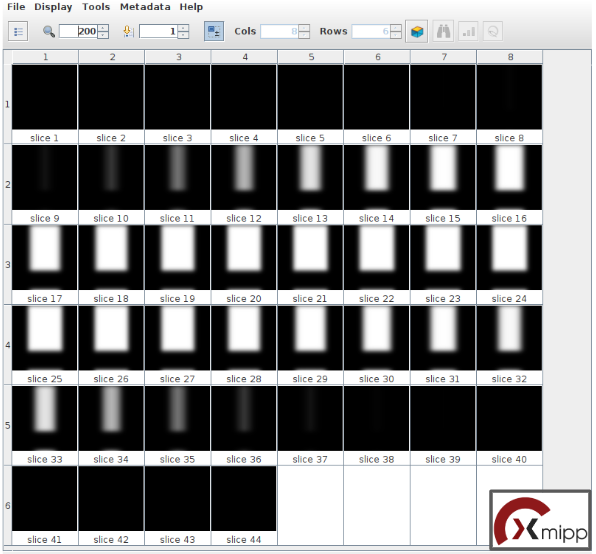
9. In the tab “Postprocessing” with the default values, set the parameter “Smooth borders” to “Yes” and “Gaussian sigma” to 2 px. This smoothing is very useful to minimize border effects.
The obtained mask, displayed in Y positive view with viewer DataViewer from xmipp, should look like shown in the figure below. To change the view, click on the colored cube ico on the top toolbar.
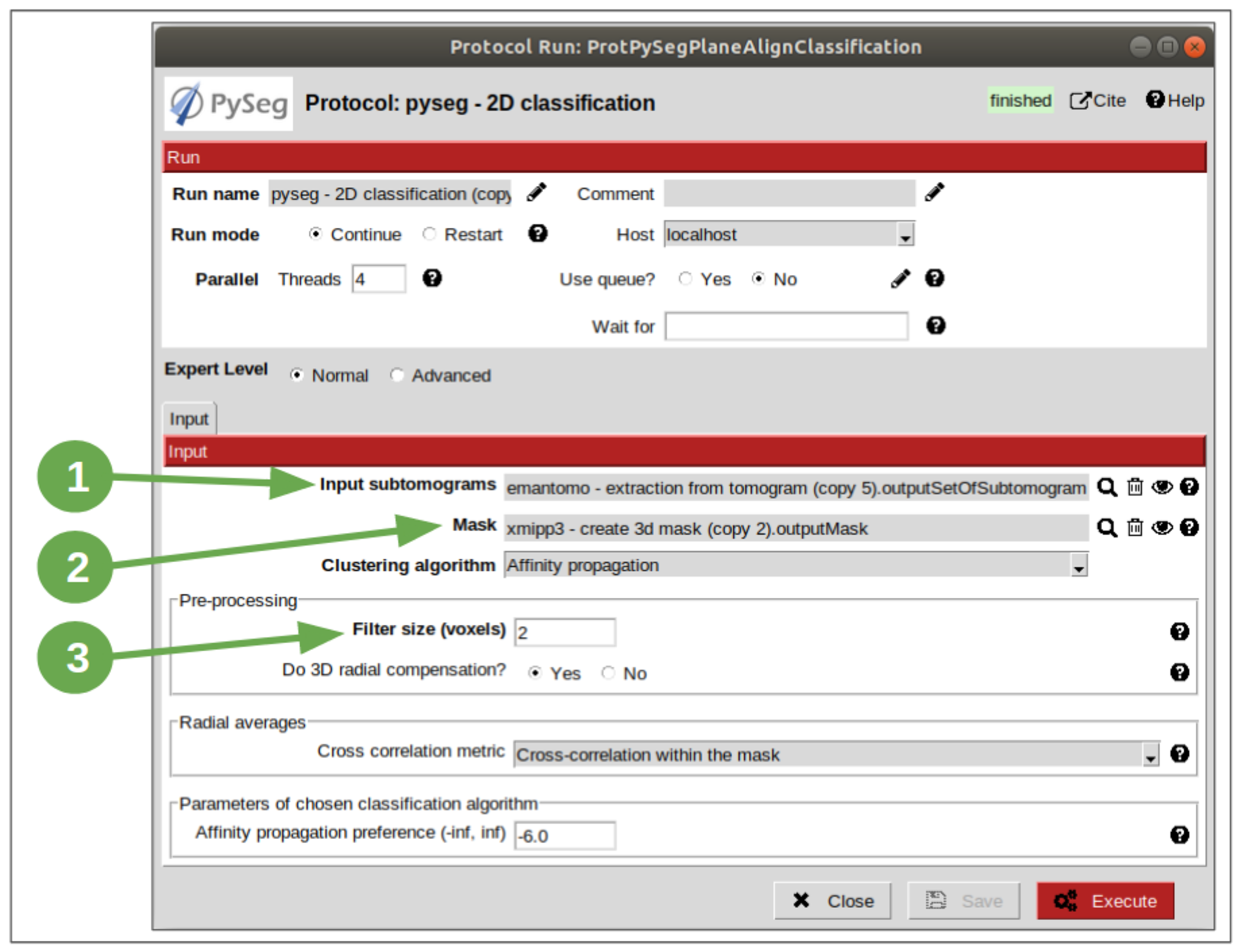
Finally, we have all the elements required to perform the 2D classification. So let’s open the protocol and set the values enumerated below:
- Set the input subtomograms pointer to the ones extracted with scipion-em-emantomo after having filtered by normal.
- Set the mask pointer to the mask generated before.
- Set the Filter size to 2 voxels.
Let all the rest of parameters with the default values. It’s remarkable that this protocol offers three different clustering algorithm, each with its own parameter, which will be shown in the protocol form when a different algorithm is selected. We’ve chosen Affinity Propagation (AP) for this tutorial due to its simplicity (number of clusters doesn’t have to be specified like in other clustering algorithms), general applicability and performance.
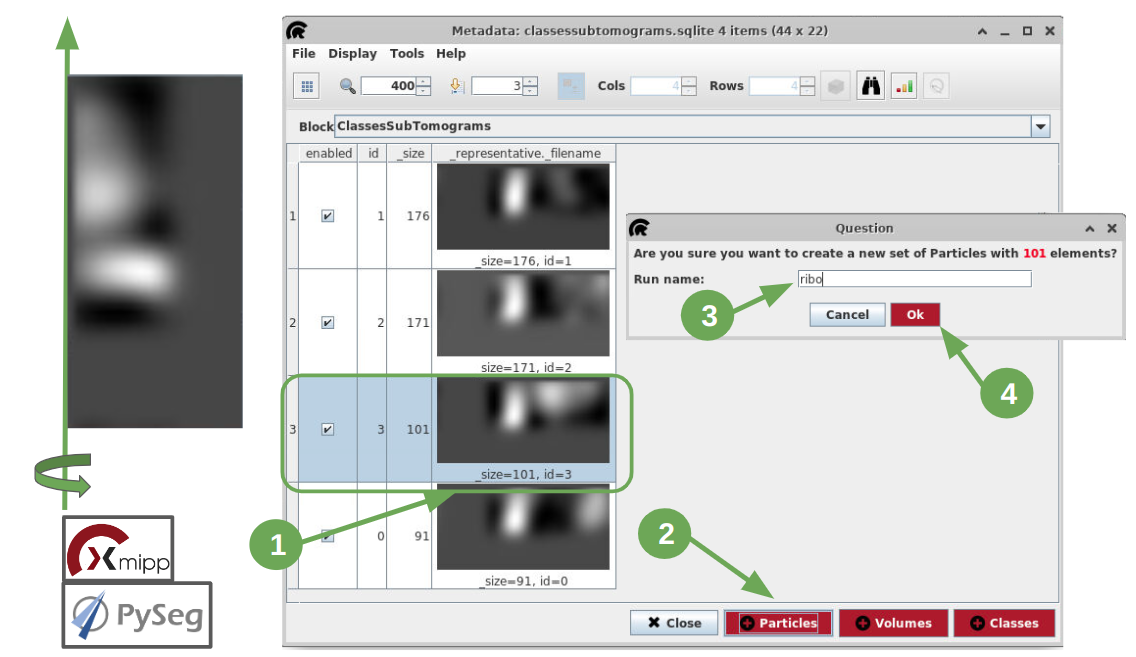
Once the protocol execution is finished, let’s right click on the outputClasses object located in the lower panel of the project interface, in the tab “Summary”. Then select the option “Open with TomoDataViewer” and the classes obtained will be displayed with xmipp’s viewer. On the top toolbar, set the size (besides the magnifier icon) to 650 and press Intro. The 2 classes obtained are represented as the rotational average around the normal axis, as can be observed on the left side of the figure below. It seems quite clear that our ribosome is on class 1, and it’s composed of 99 particles, while the other class only shows the membrane. Thus, will use the subset functionality provided by this viewer to create a subset only composed of the particles which belong to class 1:
- Select the row corresponding to class 1.
- Click on the button “+ Particles”.
- On the auxiliary window generated, choose a name for the subset. In our case it will be class1.:align:
- Click on the button “Ok” and the subset will be automatically generated.
At this point, the viewer can be closed.
SUMMARY:
That was the last point of this tutorial. If we perform some subtomogram averaging (STA) steps membrane alignment, particle alignment and subtomogram reconstruction), we can obtain a structure for our ribosomes, as shown in the figure below. Those STA steps are out of the scope of this tutorials, but the protocols used and the values of the parameters can be observed in section extra material .